Even if the implementation cited above is using the Connection Machine hardware with extremely high efficiency, there is an algorithmic problem: When forces are computed between atom pairs in neighboring grid-cells, many of the forces are in fact computed for pairs whose interatomic distance is greater than the cutoff-radius of the interactions.
A kind of ``algorithmic efficiency'' can be estimated as the number of
``relevant'' non-zero interactions compared to the total number
of calculated interactions as follows: If the cutoff-radius (or rather
the radius at which the smooth cutoff-function employed in the EMT is
considered to be effectively zero) is equal to the grid-cell side
length D, the volume of the sphere of non-zero interactions around a
given atom is  , whereas the interactions are
computed inside a volume
, whereas the interactions are
computed inside a volume  for local
for local  interaction
subgrids. The ratio of these volumes, which is an estimate of the
algorithmic efficiency of the force computation, is approximately
0.155. If the code uses local
interaction
subgrids. The ratio of these volumes, which is an estimate of the
algorithmic efficiency of the force computation, is approximately
0.155. If the code uses local  subgrids and a cutoff-radius of
subgrids and a cutoff-radius of
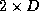 , the algorithmic efficiency is a much better 0.268. We
implemented[4] a skipping of some cells in the
, the algorithmic efficiency is a much better 0.268. We
implemented[4] a skipping of some cells in the 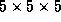 subgrid,
leading to about 20% improvement of this algorithmic efficiency.
Larger local subgrids could be used, at the cost of more communication
and a decreased number of atoms per cell. This would lead to decreased
load-balance, however, so there is an upper limit to proceeding along
this line.
subgrid,
leading to about 20% improvement of this algorithmic efficiency.
Larger local subgrids could be used, at the cost of more communication
and a decreased number of atoms per cell. This would lead to decreased
load-balance, however, so there is an upper limit to proceeding along
this line.
Thus we conclude that a fairly low upper bound to the algorithmic
efficiency of using local  subgrids exists, when
all interactions are computed. One possible resolution of this
low efficiency is to use the classical Verlet neighbor-list
method[5], but in the context of a parallel algorithm. One
implementation of such an algorithm by Tamayo is discussed in ref.
[3] (section 5).
subgrids exists, when
all interactions are computed. One possible resolution of this
low efficiency is to use the classical Verlet neighbor-list
method[5], but in the context of a parallel algorithm. One
implementation of such an algorithm by Tamayo is discussed in ref.
[3] (section 5).
We have implemented a standard Verlet neighbor-list algorithm in the context of our previously described parallel MD algorithm[4], with the aim of achieving the fastest possible execution time at the expense of using large amounts of RAM memory for storing neighbor-list related data in each parallel node.
Thus we are explicitly not aiming for simulating the largest possible systems, but rather on the fastest solution of more modest-sized systems (of the order of 1 million atoms). This tradeoff was decided in view of the physics that we want to investigate. Another aspect that influences algorithmic choices is the fact that our EMT interactions, which are more appropriate for simulating metallic systems, require more than 10 times the number of floating-point operations per pair-interaction, than for the case of Lennard-Jones interactions which are usually employed in MD studies. Hence communications overheads in our algorithm will be intrinsically much less significant than for the simpler Lennard-Jones interactions.
The implementation of a Verlet neighbor-list algorithm in a
data-parallel CM-Fortran MD code is relatively straightforward. The
task that distinguishes the data-parallel implementation from the usual
serial one is the access to data in non-local processors. Our choice
is to simply copy the data (atomic  coordinates, plus
some interaction data) from the nearby
coordinates, plus
some interaction data) from the nearby  local subgrid into the
memory of the local processor. The CSHIFT Fortran-90 intrinsic
is used for copying the data. This task is sketched in Fig. 1, and it
is carried out whenever the atoms are moved.
local subgrid into the
memory of the local processor. The CSHIFT Fortran-90 intrinsic
is used for copying the data. This task is sketched in Fig. 1, and it
is carried out whenever the atoms are moved.

Of course, a smaller part of the data will not represent neighbors of any atom in the central cell of Fig. 1, and hence this data could in principle be omitted. Also, it can be argued that it is a waste of memory to replicate atomic data from 26 neighboring cells. This is a deliberate choice that we have made: We want the most regular (and hence the fastest) communication pattern of data between the ``vector-unit'' memory of the CM-5E nodes. We also want all atomic data in a linear list in memory so that a Verlet neighbor-list algorithm can be implemented simply and efficiently. Our aim is to get the fastest possible execution at the cost of using extra memory. Since the 128-node CM-5E machine at our disposal has 128 MB of memory per node, and we want to simulate systems of only up to 1 million atoms, we can easily afford to use memory so freely.
With all relevant data available in the local memory of each node, there remains to construct the neighbor lists for each atom. Owing to all the atomic data from the central and neighboring cells being arranged in a simple linear list in local memory, it is a simple matter to traverse the list in a data-parallel fashion for each atom in the central cell and add any neighbors to a list of pointers (the neighbor-list). In CM-Fortran the atom-atom distance is calculated by a FORALL statement, and the decision whether to include the atom in the neighbor-list is implemented by a MERGE Fortran-90 intrinsic function. The pointers to atoms are added to the list by a ``scatter'' operation, which was first coded as a FORALL statement, but since the present CM-Fortran compiler (version CM5 VecUnit 2.2.11) hasn't implemented indirect memory access efficiently for 3-D arrays, we decided to use the faster CM-Fortran library CMF_ASET_1D routine in stead.
The performance-critical part of the above construction is the ``scatter'' operation, which requires local indirect-memory addressing that is emulated in software on the CM-200, but which has direct hardware support on the CM-5, so that the CM-5 can execute the neighbor-list construction exclusively with efficient vector operations on local memory. For the CM-200 the indirect-memory addressing was found to be too slow for a production code, so the present algorithm wasn't pursued any further on this hardware.
Since the CM-Fortran compiler generated sub-optimal code for local indirect-memory addressing, the CM-Fortran neighbor-list loop (a total of 7 Fortran statements) was translated into the CM-5 vector-unit assembler ``CDPEAC'', taking optimal advantage of indirect-memory addressing instructions and overlapping of computation and load/store operations. This resulted in a speedup of the neighbor-list loop by approximately a factor of 4. We also experimented with CM-Fortran ``array aliasing'' in order to aid the compiler's code generation, and while improvements were seen, the CDPEAC code was still more than twice as fast as the compiler's code.
After this optimization, an update of the neighbor-lists consumes a time roughly equal to the time for a single MD timestep. This means that the classical trick of increasing the cutoff-radius by adding a skin radius must be used in order to re-use the neighbor-list during several MD timesteps. We monitor exactly the atoms' deviations from their positions at the time of the neighbor-list construction, and when the maximum deviation exceeds half of the skin radius, an update of the neighbor-list is carried out. We believe that it is important not to postpone updates beyond this point, since the MD simulations would then not conserve energy. Atoms will migrate between cells during the simulation, and such migration-updates are performed prior to the neighbor-list updates.
When atomic forces are calculated and summed in a loop over the atoms in the neighbor-list, a simple parallel ``gather'' operation of the neighbor-atoms' data is performed using FORALL on the data structure sketched in Fig. 1. When the atomic data are in CM-5 vector-registers, the forces can be calculated by the EMT method as in ref. [4]. Again, the efficiency of the force computation depends partially on the compiler-generated code for indirect-memory access, but less so than for the neighbor-list generation because many floating-point operations are executed for each ``gather'' operation. We have considered CDPEAC coding of the ``gather'' operation, but so far this hasn't been done.